Pulmonary Langerhans Cell Histiocytosis in a young non-smoker – a rare case report.
Mahajan PA1*, Anand P2, Mullick S3
DOI:https://doi.org/10.17511/jopm.2024.i02.04
1* Pooja Awasthi Mahajan, Senior Resident, National Institute of Tb and Respiratory Diseases, New Delhi, Delhi, India.
2 Priyanshi Anand, Senior Resident, National Institute of TB and Respiratory Diseases, New Delhi, Delhi, India.
3 Shalini Mullick, Professor and HOD, National Institute of TB and Respiratory Diseases, New Delhi, Delhi, India.
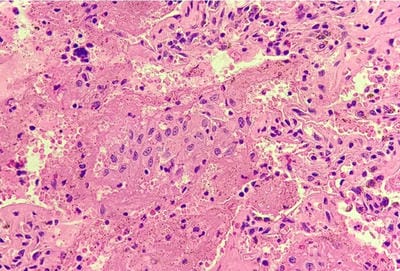
Pulmonary Langerhans cell histiocytosis (PLCH) is a diffuse cystic lung disease strongly associated with exposure to cigarette smoke. In recent times, activating pathogenic mutations in the MAPK (mitogen-activated protein kinase) pathway have been described in the dendritic cells in patients with PLCH. The outcome and disease course of PLCH are highly variable. Smoking cessation is the mainstay of treatment and can lead to disease regression or stabilization in a large percentage of patients. Further, the study of molecular pathogenesis of PLCH has anteceded the development of disease-specific biomarkers and targeted treatment options.
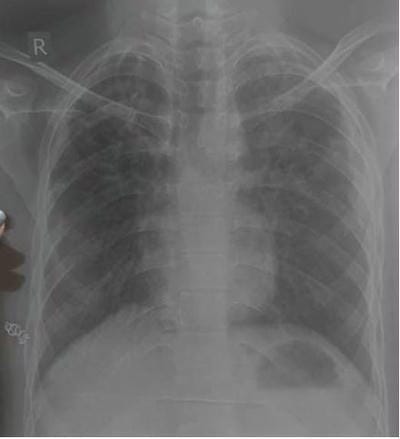
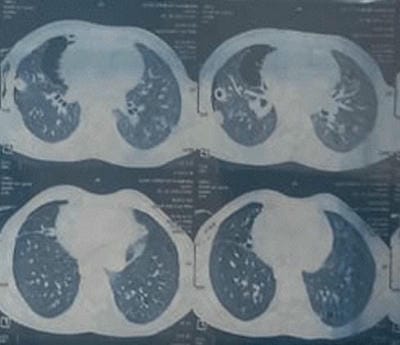
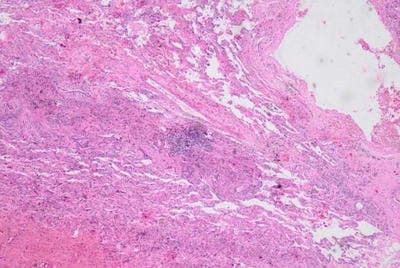
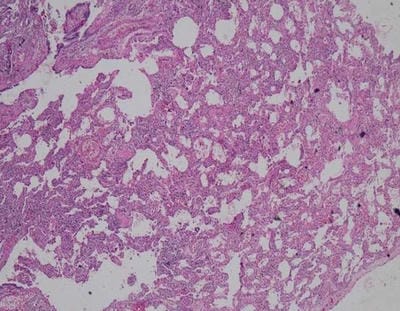
We present an interesting case report of a young non-smoker, asthmatic male with a definitive diagnosis of PLCH, clinically and radiologically. However, the classic features of Langerhans cell histiocytosis (LCH) were not seen in histopathology. We wish to highlight that advanced cases of LCH may not always have a textbook histological picture. In the appropriate clinical and radiological setting, the pathologist must consider the possibility of LCH even if classic Langerhans cells are not seen. In this paper, we would like to present the features of LCH, in the advanced stage without classic presentation.
Keywords: Imaging, MAPK, Nodules, Pulmonary Langerhans’ cell histiocytosis, Smoking
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Senior Resident, , National Institute of Tb and Respiratory Diseases, New Delhi, Delhi, India. Email:  |
Mahajan PA, Anand P, Mullick S, Pulmonary Langerhans Cell Histiocytosis in a young non-smoker – a rare case report.. Trop J Pathol Microbiol. 2024;10(2):20-23. Available From https://pathology.medresearch.in/index.php/jopm/article/view/650 |
 |
 ©
©