An Unusual Case of Residual Follicular Lymphoma with Hemophagocytic Lymphohistiocytosis
Tejesh M1, K Padma S2*, Apoorva K3, Shetty K J4
DOI: https://doi.org/10.17511/jopm.2023.i03.02
1 M Tejesh, Postgraduate, Department of Pathology, K S Hegde Medical Academy, Mangalore, Karnataka, India.
2* Shetty K Padma, Professor, Department of Pathology, K S Hegde Medical Academy, Mangalore, Karnataka, India.
3 K Apoorva, Assistant Professor, Department of Pathology, K S Hegde Medical Academy, Mangalore, Karnataka, India.
4 Jayaprakash Shetty K, Professor and Head, Department of Pathology, K S Hegde Medical Academy, Mangalore, Karnataka, India.
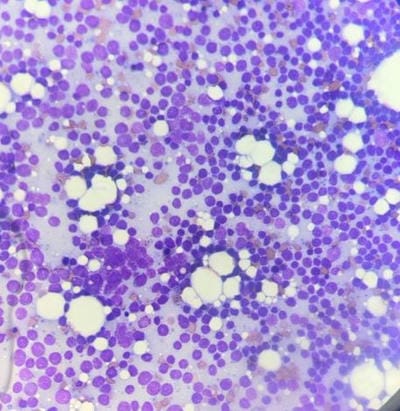
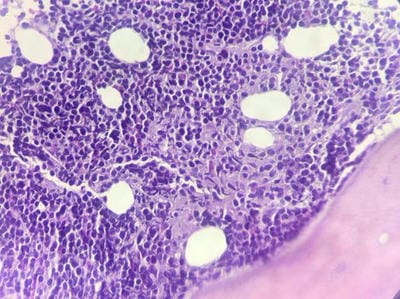
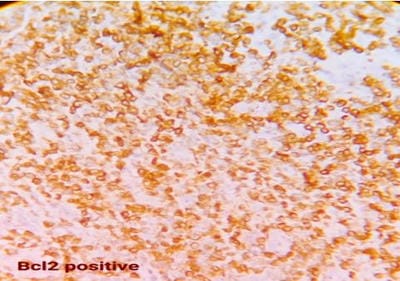
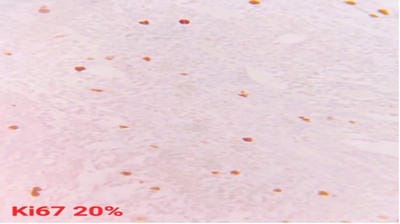
Follicular lymphoma is an indolent B-cell lymphoproliferative disorder of transformed follicular centre B cells characterised by diffuse lymphadenopathy, bone marrow involvement and splenomegaly. An uncommon, fatal clinical disease known as hemophagocytic lymphohistiocytosis is marked by hyperinflammation. HLH is brought on by abnormally activated macrophages and cytotoxic T cells, which produce acute organ dysfunction and cytokine storm. HLH caused by lymphoma is a rare but devastating condition. A rapid diagnosis will aid in effective treatment. This is a rare case report highlighting the diagnosis and treatment plan of a 60-year-old female patient with residual follicular lymphoma with hemophagocytic lymphohistiocytosis.
Keywords: Follicular, Lymphoma, Hemophagocytic Lymphohistiocytosis
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Professor, Department of Pathology, K S Hegde Medical Academy, Mangalore, Karnataka, India. Email:  |
Tejesh M, K Padma S, Apoorva K, Shetty K J, An Unusual Case of Residual Follicular Lymphoma with Hemophagocytic Lymphohistiocytosis. Trop J Pathol Microbiol. 2023;9(3):28-31. Available From https://pathology.medresearch.in/index.php/jopm/article/view/625 |
 |
 ©
©