Small round cell tumor of cervical region: a case report with unusual site & course
D. Pathak V.1, Metkar G.2*, Bhide S.3, Joshi S.4
DOI: https://doi.org/10.17511/jopm.2019.i08.09
1 Vikas D. Pathak, Department of Pathology, MIMER Medical College, Pune, Maharashtra, India.
2* Gauri Metkar, Department of Pathology, MIMER Medical College, Pune, Maharashtra, India.
3 Smita Bhide, Department of Pathology, MIMER Medical College, Pune, Maharashtra, India.
4 Sneha Joshi, Department of Pathology, MIMER Medical College, Pune, Maharashtra, India.
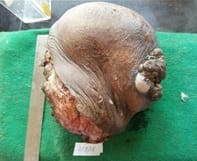
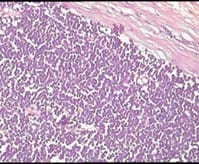
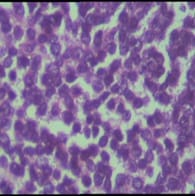
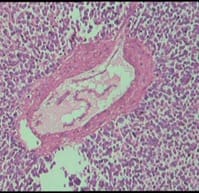
Ewing’s sarcoma belongs to a spectrum of neoplastic diseases known as Ewing’s family of tumors. Ewing sarcoma can arise in either bone or soft tissue. Small round cell tumors comprise a heterogeneous group of neoplasms that predominate in childhood and adolescence, share similar morphological features, consisting of dense cellular proliferation of small round cells with primitive appearance. Ewing sarcoma is the second most common malignant bone tumour in children and young adults, although, rarely, it may be of extraskeletal origin. Extra-osseous tumor with metastatic or recurrent disease has a worse outcome; 5-year overall survival remains about 25%. A female patient presented in surgical OPD for the first time at the age of 12 years with swelling in right supraclavicular region. The swelling was excised & a provisional diagnosis of small round cell tumor was given & patient was lost to follow up. Swelling recurred at the age of 23 years & was gradually increasing in size, measuring 20 x 20 x 20 cm. Radiologically, lesion was suggestive of sarcoma / lymphoma. Patient was operated & histopathological diagnosis of malignant round cell tumor was given with differential diagnosis of Ewing’s sarcoma, PNET, lymphoma. Patient had third recurrence of swelling at the age of 28 years at same site. On IHC, diagnosis of Soft Tissue Ewing sarcoma was given. Inspite of recurrence & huge size of tumor, patient was otherwise asymptomatic. Ours is a rare case of more than 16 years of survival inspite of recurrence.
Keywords: Small round cell tumor, Ewing sarcoma, Extra-osseous tumor, Immunohistochemistry
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Department of Pathology, MIMER Medical College, Pune, Maharashtra, India. Email:  |
Pathak VD, Metkar G, Bhide S, Joshi S. Small round cell tumor of cervical region: a case report with unusual site & course. Trop J Pathol Microbiol. 2019;5(8):562-567. Available From https://pathology.medresearch.in/index.php/jopm/article/view/310 |
 |
 ©
©